최근 우리는 인공지능이 글을 쓰고, 그림을 그리며, 심지어 코딩까지 하는 세상에 살고 있습니다. 하지만 인공지능이 ‘생명의 코드’인 DNA 를 직접 읽고, 교정하고, 심지어 무에서 유를 창조하듯 설계한다면 어떨까요? 마치 우리가 엑셀이나 워드 프로그램을 다루듯, 우리 몸의 설계도를 자유자재로 편집하는 미래 말이죠.
수조 개의 염기서열을 학습한 거대 인공지능 모델부터, 수천 년 전 인류를 괴롭혔던 질병의 유전적 기원을 밝혀낸 연구, 그리고 암 조직의 복잡한 세포 대화를 ‘도청’하는 기술까지.
오늘은 이 놀라운 성과들을 포스팅 해봅니다
유전체 기초 모델과 생성형 생물학의 시대
2026년 3월 초 Nature 지를 통해 공개된 Evo 2는 생명과학계의 ‘GPT-4’라고 불릴 만큼 압도적인 성능을 자랑합니다.[1, 2]
이 모델은 단순히 특정 유전자를 분석하는 수준을 넘어, 지구상의 모든 생명체 도메인(세균, 고균, 진핵생물)을 아우르는 방대한 유전 코드를 학습한 ‘생명체 파운데이션 모델’입니다.[1]
생명의 언어를 깨우친 9.3조 개의 뉴런, Evo 2 모델의 탄생
이전 버전인 Evo 1이 단세포 생물의 유전체에 집중했다면, Evo 2는 인간, 식물, 다세포 생물뿐만 아니라 멸종된 매머드와 같은 고대 생물의 데이터까지 포함하는 광범위한 스펙트럼을 자랑합니다.[1, 3]
이는 인공지능이 뉴클레오타이드라는 ‘생명의 알파벳’으로 이루어진 언어를 완벽하게 읽고, 쓰고, 생각할 수 있게 되었음을 의미합니다.[1]
Evo 2의 기술적 혁신과 사양
Evo 2가 가진 가장 큰 무기는 StripedHyena 2라는 독창적인 아키텍처입니다.[1]
기존의 트랜스포머(Transformer) 모델이 가진 연산 효율성의 한계를 극복하여, 한 번에 무려 100만 개의 뉴클레오타이드를 처리할 수 있는 ‘100만 토큰 컨텍스트 윈도우’를 구현했습니다.[1]
이는 유전체의 아주 먼 곳에 떨어져 있는 조절 요소들이 서로 어떻게 상호작용하는지를 한눈에 파악할 수 있게 해줍니다.
| 항목 | 상세 사양 및 특징 |
|---|---|
| 모델 명칭 | Evo 2 (7B 및 40B 파라미터 버전) [1] |
| 학습 데이터 세트 | OpenGenome2 (9.3조 개의 뉴클레오타이드) [1] |
| 학습 대상 | 10만 종 이상의 전 생명체 도메인 및 메타게놈 [1] |
| 처리 가능 길이 | 최대 100만 염기쌍 (단일 뉴클레오타이드 해상도) [2] |
| 정확도 (BRCA1 변이) | 유방암 관련 변이 예측 정확도 90% 이상 [1, 4] |
이 모델은 NVIDIA의 DGX Cloud 플랫폼에서 2,000개 이상의 H100 GPU를 사용하여 수개월 동안 훈련되었습니다. [1, 4]
연구팀은 이를 통해 AI가 단순히 서열을 외우는 것이 아니라, 진화 과정에서 축적된 생물학적 규칙과 분자 간의 상호작용 신호를 스스로 학습했다는 사실을 입증했습니다.[1, 4]
질병 예측과 맞춤형 유전자 치료의 미래
Evo 2의 실질적인 가치는 임상적 적용 가능성에서 드러납니다. 예를 들어, 유방암 유전자로 잘 알려진 BRCA1 유전자의 변이를 분석했을 때, Evo 2는 별도의 미세 조정(fine-tuning) 없이도 어떤 변이가 질병을 일으키는지 90% 이상의 확률로 정확히 가려냈습니다.[1, 4]
이는 기존의 전문 예측 도구인 AlphaMissense보다도 뛰어난 성능입니다.[1]
또한, 이 모델을 활용하면 특정 세포(예: 신경세포나 간세포)에서만 작동하는 유전자 조절 요소를 직접 설계할 수도 있습니다.[1, 4]
이는 부작용은 줄이고 효과는 극대화한 맞춤형 유전자 치료제 개발을 가속화할 수 있는 강력한 도구가 됩니다.[1]
Evo2 사용법
Evo 2는 다른 특정한 지시(작업 맞춤형 미세조정) 없이 오직 “DNA 서열만으로(from DNA sequence alone)” 해당 서열의 기능적 영향을 예측합니다.
예를 들어, 특정 유전자 서열을 입력하면 모델이 그 서열 내의 돌연변이가 무해한지 혹은 유방암(BRCA1)과 같은 질병을 유발할 수 있는 병원성을 띠는지 90% 이상의 정확도로 가려냅니다.
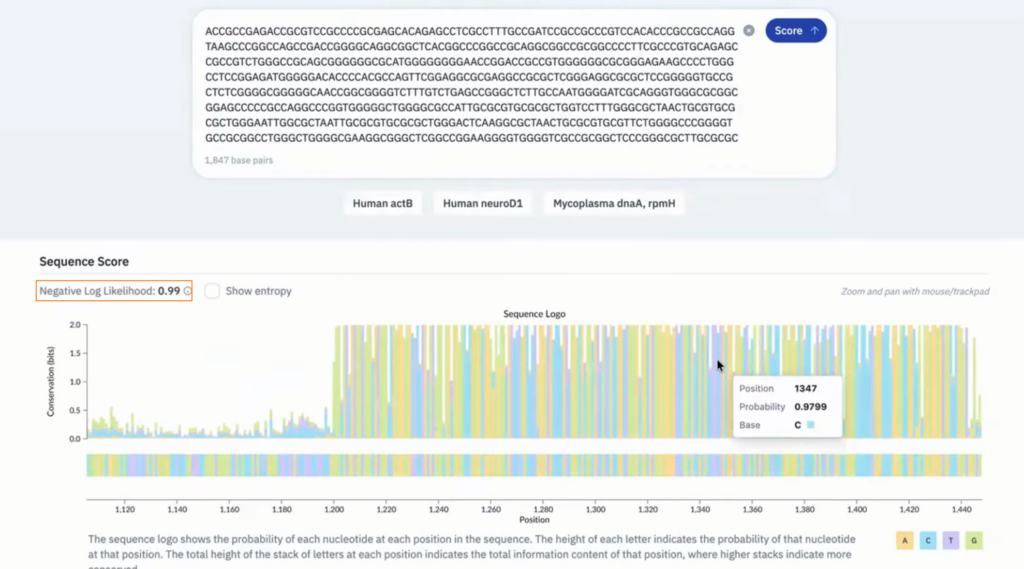
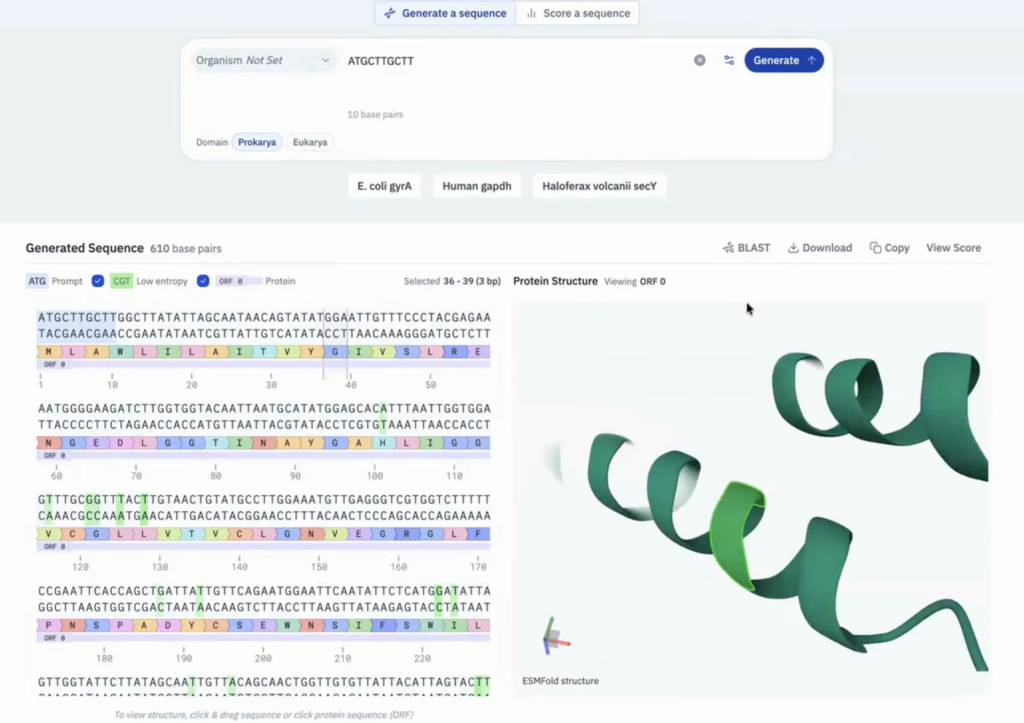
현재 Evo 2의 매개변수(파라미터), 학습 코드, 추론 코드 등은 완전히 오픈 소스로 공개되어 Arc Institute의 GitHub에서 누구나 접근할 수 있습니다.
또한, NVIDIA의 BioNeMo 프레임워크에도 통합되어 있어 연구자들이 직접 모델을 가져와 방대한 염기서열 데이터를 처리하고 분석 실험을 수행하는 데 사용할 수 있습니다
미생물학 및 항생제 내성 기전의 분자적 이해
항생제 내성 세균, 즉 슈퍼박테리아는 현대 의학이 직면한 가장 거대한 과제 중 하나입니다.
2026년 2월 26일자 Nature 지에는 박테리오파지(세균을 감염시키는 바이러스)가 어떻게 세균의 방어벽을 뚫고 나오는지에 대한 놀라운 연구 결과가 실렸습니다.[5]
슈퍼박테리아를 무너뜨리는 바이러스의 ‘킬 스위치’
칼텍(Caltech)의 빌 클레먼스(Bil Clemons) 교수 연구팀은 바이러스 단백질이 세균의 핵심 수송 단백질인 MurJ를 마비시키는 메커니즘을 규명했습니다.[5]
MurJ: 세균 성벽의 건설자
박테리아는 자신을 보호하기 위해 ‘펩티도글리칸’이라는 단단한 세포벽을 만듭니다.
이때 MurJ라는 단백질은 세포벽을 만드는 재료를 세포 안에서 밖으로 나르는 ‘회전문’ 같은 역할을 합니다.[5] 만약 이 회전문이 멈추면 세균은 더 이상 세포벽을 유지할 수 없어 결국 죽게 됩니다.[5]
| 주요 단백질 | 역할 및 기전 |
|---|---|
| MurJ | 펩티도글리칸 전구체를 막 너머로 이동시키는 플립페이즈(Flippase) [5] |
| Sgl 단백질 | 바이러스가 생성하여 MurJ의 홈에 결합, 작동을 멈추게 하는 단백질 [5] |
| MraY, MurG | 세포벽 생성 과정의 다른 필수 효소들 (새로운 약물 타깃 후보) [5] |
펩티도글리칸
Peptidoglycan(peptidoglycan)은 세균 세포벽을 구성하는 거대 고분자(polymer)로, 당(glycan) 사슬과 펩타이드(peptide) 교차결합으로 이루어진 망상 구조(mesh structure)를 가집니다.
연구팀은 극저온 전자현미경(Cryo-EM)을 통해 서로 다른 세 종류의 바이러스 단백질(SglM, SglPP7, SglCJ3)이 모두 동일한 방식으로 MurJ를 공격한다는 사실을 발견했습니다.[5]
이 바이러스들은 진화적으로 서로 연관이 없음에도 불구하고, MurJ의 특정 부위에 결합하여 이를 ‘바깥쪽을 향한 구조’로 고정해버립니다.
이는 사냥꾼들이 각자 다른 곳에서 왔지만, 결국 사냥감을 잡는 가장 효율적인 방법 하나를 똑같이 찾아낸 것과 같은 ‘수렴 진화’의 사례입니다.[5]
이 발견은 매우 중요한 의미를 가집니다. MurJ는 세균에게는 필수적이지만 인간 세포에는 존재하지 않기 때문에, 이를 공격하는 새로운 종류의 항생제를 만든다면 부작용 없이 슈퍼박테리아만 선택적으로 제거할 수 있기 때문입니다.[5]
단일 세포 및 공간 전사체 통합 분석 기술
최근 생명과학계의 화두는 ‘어떤 세포가(Single-cell)’ ‘어디에 위치하며(Spatial)’, ‘주변과 어떻게 대화하는가’를 알아내는 것입니다.
하지만 기존의 기술로는 이 세 가지 정보를 한꺼번에 완벽하게 얻기 어려웠습니다. 2026년 2월 Bioinformatics 지에 발표된 transFusion 플랫폼은 이 복잡한 퍼즐 조각들을 하나로 합쳐주는 혁신적인 웹 기반 분석 도구입니다.[6, 7]
조직의 지도를 그리는 새로운 도구, transFusion
기존의 10x Visium 공간 전사체 기술은 해상도가 약 55마이크로미터로, 하나의 점(Spot) 안에 여러 종류의 세포가 섞여 있어 정확한 분석이 어려웠습니다.
transFusion은 단일 세포 RNA 시퀀싱(scRNA-seq) 데이터를 결합하여 각 지점에 어떤 세포가 어떤 비율로 섞여 있는지 정확히 추정(Deconvolution)해냅니다.[6, 8]
| 주요 기능 | 설명 및 기대 효과 |
|---|---|
| 세포 유형 역분해 | 혼합된 데이터에서 개별 세포의 비율을 계산하여 해상도 보완 [6] |
| 리간드-수용체 상호작용 | 세포 간 신호 전달 분자를 식별하여 세포 대화 분석 [9] |
| 공간적 의존성 분석 | 특정 세포들이 왜 항상 붙어 있는지, 어떤 영향을 주는지 파악 [8] |
| 다중 양식 기울기 분석 | 암 조직의 경계 등에서 발생하는 점진적인 유전자 변화 탐지 [8] |
실제로 이 플랫폼은 유방암 조직 분석에서 암세포와 주변 섬유아세포(CAF), 면역세포 사이의 복잡한 통신망을 밝혀냈습니다.[6]
예를 들어, MIF-CD74나 FN1-ITGA5와 같은 특정 신호 전달 경로가 암의 진행에 어떻게 기여하는지를 공간적인 맥락에서 시각화해준 것입니다.[6]
고고유전학: 유전체로 다시 쓰는 인류의 역사
2026년 2월 Science 지에는 마치 인디아나 존스 영화 같은 흥미로운 연구가 발표되었습니다.
콜롬비아 보고타(Sabana de Bogotá) 지역에서 발견된 5500년 전의 인골에서 매독균(Treponema pallidum)의 유전체를 성공적으로 복원하고 분석한 연구입니다.[12]
5500년의 침묵을 깬 매독균의 유전체
수천 년 된 유전체(Ancient DNA)는 현대의 DNA와 달리 매우 심하게 훼손되어 있고, 주변 토양의 미생물 DNA와 섞여 있어 분석이 매우 까다롭습니다.
연구진은 이를 해결하기 위해 정교한 생명정보학 파이프라인을 구축했습니다.
- Authentication(인증): DamageProfiler 도구를 사용하여 시간이 지남에 따라 발생하는 DNA의 특유한 화학적 변형(C에서 T로의 전이)을 확인하여, 이 DNA가 정말 수천 년 전의 것임을 입증했습니다.[12]
- Competitive Mapping: 매독균과 유사한 다른 균들과 서열을 일일이 대조하여, 오직 매독균 계통의 서열만을 정밀하게 추출해냈습니다.[12]
- Molecular Clock(분자 시계): 유전적 변이가 축적된 속도를 바탕으로 역계산하여, 이 균이 현대의 매독균들과 언제 갈라졌는지를 추정했습니다.[12]
분석 결과, 이 5500년 전의 매독균은 현대의 모든 매독 아종들이 분화되기 이전의 초기 형태인 것으로 밝혀졌습니다.[12]
이는 인류를 수세기 동안 괴롭혀온 매독이라는 질병이 어떻게 탄생했고, 어떤 경로로 전 세계에 퍼져 나갔는지를 이해하는 데 있어 결정적인 퍼즐 조각을 제공합니다.
질병의 과거를 아는 것은 우리가 앞으로 마주할 새로운 감염병의 진화 방향을 예측하는 데에도 큰 도움이 됩니다.
의생명 텍스트 마이닝과 거대 언어 모델(LLM)의 진화
매년 수백만 편의 의생명 논문이 쏟아져 나오지만, 이를 사람이 일일이 읽고 정보를 정리하는 것은 불가능에 가깝습니다.
이를 해결하기 위해 AI가 논문에서 핵심 정보를 자동으로 추출하는 기술이 개발되고 있습니다.[13, 14, 15]
AI에게 의학 논문을 완벽하게 이해시키는 방법: BioTriplex
기존의 데이터셋들은 논문의 요약본인 ‘초록’만을 대상으로 학습하는 경우가 많아, 본문에 숨겨진 상세한 맥락이나 부정적인 결과(이 유전자는 질병과 상관없음 등)를 놓치는 경우가 많았습니다.
BioTriplex는 100편의 논문 전체 텍스트(Full-text)를 사람이 직접 읽고, 유전자와 질병 사이의 관계를 무려 21가지의 세부 유형으로 분류하여 라벨링했습니다.[14, 15]
| 특징 | 기존 데이터셋 | BioTriplex |
|---|---|---|
| 분석 범위 | 주로 논문 초록 (Abstract) | 논문 전체 텍스트 (Full-text) [14] |
| 관계 유형 | 단순 연관성 (관련 있음/없음) | 21가지의 상세한 하위 유형 분류 [13] |
| 학습 모델 | BERT 기반 소형 모델 | LLaMA 3.1 8B 등 최신 LLM [14] |
연구진은 이 고품질 데이터를 사용하여 비교적 크기가 작은 LLaMA 3.1 8B 모델을 미세 조정했습니다.
놀랍게도 이 모델은 훨씬 더 거대한 몸집을 가진 GPT-4나 Claude 3.7 Sonnet 모델보다 유전자-질병 관계 추출 작업에서 더 뛰어난 성능을 보였습니다.[13, 14]
이는 범용 인공지능보다 특화된 전문 지식을 학습한 ‘도메인 특화 AI’의 중요성을 잘 보여주는 사례입니다.
생명정보학 소프트웨어의 품질과 안정성
우리가 사용하는 스마트폰 앱에 버그가 있으면 조금 불편하고 말겠지만, 환자의 유전자를 분석하고 질병을 진단하는 생명정보학 소프트웨어에 오류가 있다면 이는 생명과 직결되는 심각한 문제가 됩니다.
2026년 2월 IEEE 관련 저널 등에서는 생명정보학 소프트웨어의 결함(Defect) 밀도를 분석한 흥미롭고도 경고 섞인 연구가 발표되었습니다.[16]
언어별 결함 밀도의 차이
연구에 따르면, 생명정보학 소프트웨어는 일반적인 상용 소프트웨어보다 결함 밀도가 평균적으로 11.8배나 높았습니다.[16]
특히 사용되는 프로그래밍 언어에 따라 그 차이가 뚜렷했습니다.
| 프로그래밍 언어 | 평균 결함 밀도 (KLOC당 개수) |
|---|---|
| R (통계 분석용) | 1202개 [16] |
| C/C++ (고성능 연산용) | 159개 [16] |
| Python (범용/AI용) | 51개 [16] |
R 언어 기반의 소프트웨어에서 유독 높은 결함이 발견된 이유는, 많은 연구자가 엄격한 소프트웨어 공학적 훈련을 받기보다는 분석 자체에 집중하여 코드를 작성하기 때문인 것으로 분석됩니다.[16]
연구진은 특히 ‘주석(Annotation)’과 관련된 오류가 많아, 추후 코드 유지보수나 보안 취약점 해결에 큰 걸림돌이 된다고 지적했습니다.
참고
- With Evo 2, AI Can Model And Design The Genetic Code For All …, https://astrobiology.com/2026/03/with-evo-2-ai-can-model-and-design-the-genetic-code-for-all-domains-of-life.html
- Genome Modeling And Design Across All Domains Of Life With Evo 2 – Astrobiology Web, https://astrobiology.com/2025/02/genome-modeling-and-design-across-all-domains-of-life-with-evo-2.html
- Generative AI tool marks a milestone in biology | Stanford Report, https://news.stanford.edu/stories/2025/02/generative-ai-tool-marks-a-milestone-in-biology-and-accelerates-the-future-of-life-sciences
- Evo 2 AI Models Genetic Code for All Life Domains – Berkeley Today, https://nationaltoday.com/us/ca/berkeley/news/2026/03/05/evo-2-ai-models-genetic-code-for-all-life-domains/
- Scientists discover a bacterial kill switch and it could change the …, https://www.sciencedaily.com/releases/2026/02/260228082723.htm
- Volume 42 Issue 2 | Bioinformatics | Oxford Academic, https://academic.oup.com/bioinformatics/issue/42/2
- transFusion: a Novel Comprehensive Platform for integration Analysis of Single-Cell and Spatial Transcriptomics | Request PDF – ResearchGate, https://www.researchgate.net/publication/400517635_transFusion_a_Novel_Comprehensive_Platform_for_integration_Analysis_of_Single-Cell_and_Spatial_Transcriptomics
- transFusion: a novel comprehensive platform for integration analysis of single-cell and spatial transcriptomics – Oxford Academic, https://academic.oup.com/bioinformatics/article/42/2/btag059/8465947
- transFusion: a novel comprehensive platform for integration analysis of single-cell and spatial transcriptomics – Oxford Academic, https://academic.oup.com/bioinformatics/advance-article-pdf/doi/10.1093/bioinformatics/btag059/66792045/btag059.pdf
- Information Geometry Reconciles Discrete and Continuous Variation in Single-Cell and Spatial Transcriptomic Analysis | bioRxiv, https://www.biorxiv.org/content/10.64898/2026.02.25.707866v1
- Information Geometry Reconciles Discrete and Continuous Variation in Single-Cell and Spatial Transcriptomic Analysis – bioRxiv.org, https://www.biorxiv.org/content/10.64898/2026.02.25.707866v1.full.pdf
- Scientific publications 2026 | SIB Swiss Institute of Bioinformatics, https://www.sib.swiss/community/publications/scientific-publications-2026
- BioTriplex: A Full-Text Annotated Corpus for Fine-Tuning Language Models in Gene-Disease Relation Extraction Tasks | Request PDF – ResearchGate, https://www.researchgate.net/publication/399965375_BioTriplex_A_Full-Text_Annotated_Corpus_for_Fine-Tuning_Language_Models_in_Gene-Disease_Relation_Extraction_Tasks
- BioTriplex: a full-text annotated corpus for fine-tuning language models in gene-disease relation extraction tasks – Oxford Academic, https://academic.oup.com/bioinformatics/article/42/2/btag037/8435810
- [PDF] TBGA: a large-scale Gene-Disease Association dataset for Biomedical Relation Extraction | Semantic Scholar, https://www.semanticscholar.org/paper/36812ad807a493aac6143d1f8fc7aea16992de94
- Reaching Software Quality for Bioinformatics Applications: How Far Are We?, https://www.computer.org/csdl/journal/ts/2026/02/11281877/2ckbODhxe2A
- RCSB PDB: Homepage, https://www.rcsb.org/
- Special Issue : Emerging Trends in Bioinformatics and Computational Biology – MDPI, https://www.mdpi.com/journal/cimb/special_issues/Z63S09KJ8Q
- BioMedInformatics, Volume 6, Issue 1 (February 2026) – 9 articles, https://www.mdpi.com/2673-7426/6/1
- Cells, Volume 15, Issue 3 (February-1 2026) – 100 articles – MDPI, https://www.mdpi.com/2073-4409/15/3
- Advance articles | Genomics, Proteomics & Bioinformatics – Oxford Academic, https://academic.oup.com/gpb/advance-articles
- An expanded role for single-cell chemical genomics profiling in drug discovery | Biochemical Journal | Portland Press, https://portlandpress.com/biochemj/article/doi/10.1042/BCJ20253273/237111/An-expanded-role-for-single-cell-chemical-genomics
- Lipid Interactome: an interactive and open access platform for exploring cellular lipid–protein interactions | Bioinformatics | Oxford Academic, https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/btaf651/8489746?searchresult=1
- PICNIC web server for predicting proteins involved in biomolecular condensates | Bioinformatics | Oxford Academic, https://academic.oup.com/bioinformatics/article/42/1/btaf647/8362260